AI Plays Tetris with Proteins
Pioneering the Future of Drug Discovery with Artificial Intelligence
Discover MoreWhat We Do
An AI-Powered Preclinical Drug Discovery Company
Pharma R&D efficiency is decreasing steadily, while the cost of new drug discovery is increasing exponentially. Vast amounts of structured data are being generated using high throughput technologies in drug discovery/development. However, the analysis and interpretation of this data for meaningful outcomes has been a challenge.
Peptris has developed a platform technology to enhance efficiencies across the drug discovery/development cascade using Artificial Intelligence/Machine Learning.
Peptris’ proprietary AI platform integrates unsupervised learning and generative AI to design optimized molecules, significantly reducing the need for wet lab testing. The automated platform streamlines this process, designing potent, safe, selective, and biologically active drug candidates. The platform has been used to discover Novel Chemical Entities (NCE), to repurpose or reposition approved drugs, and rescue molecules that have been proven safe in the clinic. We have a pipeline of preclinical assets that have been discovered, validated in vitro and in vivo disease models.
Peptris operates virtually, focusing on AI-driven discovery while outsourcing wet lab work to CROs and biotech partners. Revenue comes from licensing deals, milestone payments, and royalties, with partnerships across academia, biotech and pharma companies and rare disease organizations.
Novel Chemical Entity
Generate novel scaffolds and molecules that can be synthesised, with superior activity, selectivity and optimal drug like properties.
Repurpose Approved Drugs
Repurpose approved drugs and molecules with proven clinical safety to find solutions for unmet medical needs and rare disease indications.
Rescue Clinical Candidates
Identify novel molecular targets and disease indications for clinically safe molecules that have passed Phase I or later stages.
Technology
We Accelerate Drug Discovery
Our proprietary technology leverages cutting edge neural network architectures found in Natural Language Processing, Large Language Models and Image processing research areas.
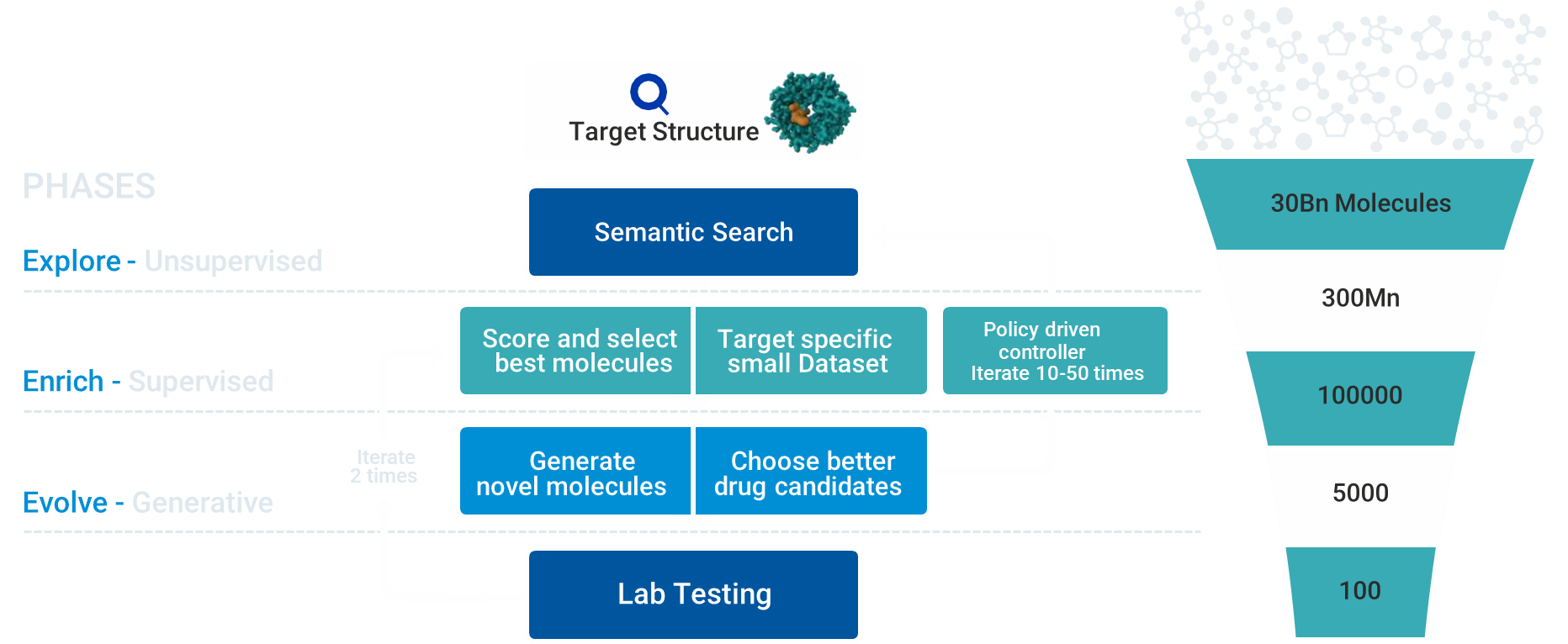
Peptris has created an array of AI models to understand the chemical space and generate novel molecules and predict varied parameters that are critical for a molecule to be considered a potential drug candidate. The power of the Peptris platform is derived from a foundational unsupervised learning algorithm to understand the vast chemical space. It uses a large language model built on a novel and proprietary molecule language syntax.
Our proprietary generative AI algorithms can also design novel molecules with superior physicochemical properties and drug target activity. The models are orchestrated by an automated process that prunes large purchasable compound datasets and creates de novo variations of compounds that are predicted to be novel, potent, safe, selective, and biologically active.
Features
The Peptris Advantage
Novel
Generates diverse, patentable and synthesizable molecules.
Fast
Rapidly screens ultra-large diverse virtual chemical libraries.
Accurate
Predicts critical parameters with high accuracy.
Cost Effective
Reduces the number of molecules for wet lab testing.
Our Progress
Discovery Pipeline
Novel Molecules
Repurposed Drugs
Portfolio
Strategic Partnerships & Success Stories

A Landmark Deal: PEPR-124 for Duchenne Muscular Dystrophy
In a significant milestone for AI-driven drug discovery in India, Peptris has entered into an exclusive licensing agreement with Revio Therapeutics. This partnership will advance our lead asset, PEPR-124, for the treatment of Duchenne Muscular Dystrophy (DMD), a rare genetic disorder with high unmet medical needs.
-
Licensed Asset: PEPR-124 (RT-001), a repurposed, Phase 2-ready candidate.
-
Target Indication: Duchenne Muscular Dystrophy (DMD).
-
Regulatory Status: Granted Orphan Drug Designation (ODD) by the USFDA.
-
Key Advantage: A mutation-agnostic therapeutic with a strong safety profile, discovered using our proprietary AI platform.
About Us
Our Team of Innovators
Founding Team

Venkatasubramanian Narayanan
Chief Executive Officer
Specialist in Algorithms. Expertise in building large systems and software. Keen Observer and a great leader.

Anand Budni
Chief Technology Officer
Expert in Image and Video processing and Deep Learning Architectures. The creative thinker and eternal optimist.


Core Team







Our Updates
News and Media
Contact Us
Get In Touch
Our Location
Center for Cellular and Molecular Platforms (C-CAMP),
NCBS-TIFR Campus, GKVK, Bellary Road, Bangalore, India